Kallmann sendromu
Astrid Leitner, Viyana'da veterinerlik eğitimi aldı. Veterinerlik mesleğinde on yıl geçirdikten ve kızının doğumundan sonra - daha çok şans eseri - tıbbi gazeteciliğe geçti. Tıbbi konulara olan ilgisinin ve yazma sevgisinin onun için mükemmel bir kombinasyon olduğu kısa sürede anlaşıldı. Astrid Leitner kızı, köpeği ve kedisi ile Viyana ve Yukarı Avusturya'da yaşıyor.
houseofgoldhealthproducts uzmanları hakkında daha fazla bilgi Tüm içeriği tıp gazetecileri tarafından kontrol edilir.Kallmann sendromu, seks hormonlarının çok az üretildiği veya hiç üretilmediği doğuştan gelen bir hastalıktır. Etkilenenler ergenliğe girmez; tedavi edilmezse hem erkek hem de kadınlar kısırdır. Ayrıca koku alma duyusu da genellikle bozulur: KS hastaları çok az koku alır ya da hiç kokmaz. Olfaktogenital sendrom hakkında bilmeniz gereken her şeyi buradan okuyun!
Bu hastalık için ICD kodları: ICD kodları, tıbbi teşhisler için uluslararası kabul görmüş kodlardır. Örneğin, doktor mektuplarında veya iş göremezlik belgelerinde bulunabilirler. E23

Kısa bir bakış
- Kallmann Sendromu nedir? Cinsiyet hormonlarının eksikliğine ve dolayısıyla ergenliğin yokluğuna yol açan doğuştan gelişimsel bozukluk. Ayrıca hastaların çoğunda koku alma duyusu yoktur.
- Nedenleri: Konjenital genetik değişiklikler (mutasyonlar)
- Risk faktörleri: Hastalık, hastaların yaklaşık yüzde 30'unda ailelerde görülür.
- Semptomlar: Ergenlik gelişiminin olmaması (penis, testisler ve prostatın az gelişmiş olması, kasık, koltuk altı ve vücut kıllarının az olması, sakalın çıkmaması, ilk adet döneminin olmaması), kısırlık, haz alamama, koku alma duyusu yok veya çok fazla azaltılmış, uzun vadeli sonuç: osteoporoz
- Teşhis: Sekonder cinsel özelliklerin gelişmemesi, hormon analizi, genetik test, ultrason, manyetik rezonans tomografi, bilgisayarlı tomografi, röntgen gibi fiziksel belirtiler
- Tedavi: hormonal ilaçlarla replasman tedavisi
- Önleme: Önleme mümkün değil
Kallmann Sendromu nedir?
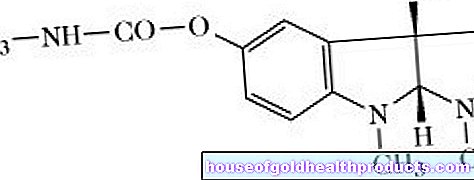
Kallmann sendromu (KS, olfaktogenital sendrom, De-Morsier-Kallmann sendromu) beynin doğuştan gelişimsel bir bozukluğudur. Etkilenenlerin seks hormonları üretmemesini ve dolayısıyla cinsel olgunluk oluşmamasını sağlar. Ek olarak, çoğu KS hastası anosmiden muzdariptir, bu da herhangi bir koku algılamadıkları anlamına gelir.
Hastalığın nedeni, anne karnındaki embriyonik gelişimi zaten etkileyen bir genetik değişimdir (mutasyon). Beynin hormonal kontrol merkezinde (hipotalamus), daha sonraki yaşamda seks hormonu üretiminin ağır basan kontrolünden sorumlu olan bazı hücreler eksiktir. Ek olarak, gen modifikasyonu, beyindeki koku alma merkezinin (olfaktör ampul) gelişmediği veya tam olarak gelişmediği anlamına gelir. Etkilenen insanlar hiçbir şey kokmazlar veya çok sınırlı bir oranda koku alırlar.
Hastalık adını 1944 yılında "hipogonadizm (seks hormonlarının eksikliği) ve anosmi (koku alma duyusu eksikliği)" olarak tanımlayan Alman psikiyatrist Franz Josef Kallmann'dan almıştır. Eşanlamlı “De Morsier-Kallmann Sendromu” terimi, aynı zamanda Kallmann sendromu üzerinde çalışmalar yürüten İsviçreli nörolog Georges de Morsier'i de ifade eder.
ikincil hipogonadizm
Kallmann sendromu, ikincil hipogonadizmin bir alt formudur. Bu, beyindeki hormonal kontrol merkezindeki (hipofiz bezi) bir arıza tarafından tetiklenen seks hormonlarının eksikliği anlamına gelir. Gonadlar (yumurtalıklar, testisler) normal çalışır, ancak seks hormonlarının üretimi hakkında çok az sinyal alır veya hiç almaz. Hormon eksikliğine ek olarak anosmi (koku alma eksikliği) varsa doktorlar Kallmann sendromundan bahseder.
Sıklık
Kallmann sendromu nadir görülen bir hastalıktır; erkekler kadınlardan daha sık etkilenir: ortalama olarak 10.000 erkekten birinde ve 50.000 kadından birinde görülür.
Belirtiler
Kallmann sendromunun ana semptomları, büyük ölçüde azalmış veya koku alma duyusunun olmaması ve ergenlik gelişiminin olmamasıdır. Semptomların ne kadar şiddetli olduğu hastadan hastaya değişir. Tedavi edilmezse her iki cinsiyet de kısır kalır.
Eksik ergenlik: Cinsiyet hormonları, ergenliğin başlangıcı için gereklidir. Çok az veya hiç seks hormonu yoksa, ergenlik ve dolayısıyla ikincil cinsel özelliklerin gelişimi gerçekleşmez.
Erkek çocuklarda belirtiler:
- Kasık, koltuk altı ve vücut kıllarının az olması veya hiç olmaması
- eksik sakal
- Sesini kırmamak
- Mikropenis ve küçük testisler: Genç erkeklerde penis ve testisler gelişmez.
- İnmemiş testisler (genellikle bebeklik döneminde belirgindir)
- Uzun boy: Sağlıklı kişilerde uzun kemiklerdeki büyüme plakları belli bir seks hormonu düzeyine ulaşılır ulaşılmaz kapanır. Hormonlar eksikse, etkilenenler orantısız bir şekilde büyür. Ebeveynlerinden daha uzundurlar ve uzun kolları ve bacakları vardır.
Kızlarda belirtiler:
Kızlarda semptomlar genellikle daha az belirgindir. Genellikle tek semptom, ilk adet kanamasının olmamasıdır (birincil amenore), bu nedenle hastalık genellikle geç fark edilir. Erkek çocukların aksine, hastalığa rağmen fiziksel gelişim büyük ölçüde normaldir. Örneğin, göğüsler genellikle normal olarak gelişir.
Erkek erişkinlerde belirtiler:
Kallmann sendromlu erkeklerde tipik olarak kemik yoğunluğu (osteoporoz) ve kas kütlesi azalır. Genellikle feminen bir yağ dağılımı nedeniyle feminen bir dış görünüme sahiptirler. Erektil disfonksiyonun var ve kısırsın.
Koku alma duyusunda azalma veya yokluk: KS hastaları kokuları algılamazlar (anosmi) veya sadece kokuları zayıf algılarlar (hiposmi). Bu belirti genellikle fark edilmez çünkü insanlar doğuştan hiçbir koku almamaya alışmışlardır. Koku alma duyusunun olmaması, münferit durumlarda, örneğin etkilenenler yanık kokusunu veya bozulmuş gıda kokusunu algılamazlarsa, tehlikeli durumlara yol açabilir.
Malformasyonlar: Diğer fiziksel malformasyonlar KS hastalarında daha az sıklıkla görülür. Bunlar örneğin işitme bozuklukları, yarık dudak, çene ve damak veya diş eksikliğidir. Bazı insanlar tek böbrekle doğarlar. KS'de zihinsel gelişim genellikle normaldir.
Osteoporoz: Testosteron ve östrojen gibi seks hormonları, kemik mineralizasyonunda önemli bir rol oynar. Hormonlar eksikse, kemikler sağlıklı insanlarda olduğu kadar stabil değildir - kemik kırılma riski artar.
Sebep ve risk faktörleri
Kallmann sendromunun nedeni doğuştan gelen bir genetik değişikliktir (mutasyon).Genellikle kendiliğinden ortaya çıkar, vakaların yaklaşık yüzde 30'unda bir veya iki ebeveynden yavrulara geçer.
Genetik değişim, rahimdeki embriyonik gelişimi zaten etkiler: koku alma hücreleri ve gonadları (yumurtalıklar, testisler) kontrol etmekten sorumlu hücreler, ortak progenitör hücrelerden kaynaklanır.
Kallmann sendromunda, bu progenitör hücrelerin gelişimi, genetik değişim tarafından bozulur. Sonuç olarak, seks hormonu üretiminin ağır basan kontrolünden sorumlu olan hücreler ve koku alma hücreleri yeterince gelişmemiştir. Etkilenen insanlar ergenlik geliştirmezler ve herhangi bir koku algılamazlar.
seks hormonları
Hipotalamus (diensefalondaki bir bölge) vücudun hormonal kontrol merkezidir. Sağlıklı insanlarda hipotalamus, GnRH hormonunu (gonadotropin salgılatıcı hormon) salgılar. Bu da hipofiz bezini LH (luteinize edici hormon) ve FSH (folikül uyarıcı hormon) hormonlarını salması için uyarır.
LH ve FSH, nihayetinde seks hormonlarını üreten gonadlara (yumurtalıklar, testisler) etki eder: kadınlarda, kadın cinsiyet hormonları östrojen ve progesteron, yumurta hücrelerinin olgunlaşmasını teşvik eder ve yumurtlamayı tetikler, erkeklerde testosteron hormonu oluşumuna neden olur. sperm.
Ergenlik döneminde seks hormonlarının üretimi ve bununla birlikte cinsel olgunlaşma başlar. Kallmann sendromunda olduğu gibi çok az GnRH varsa veya hiç yoksa seks hormonu çok az üretilir veya hiç üretilmez ve ergenlik gelişmez. Tedavi edilmezse, etkilenenler cinsel olgunluk yaşamazlar ve kısır kalırlar.
Risk faktörleri
Çoğu durumda, genetik değişiklik kendiliğinden gerçekleşir. Bunun nasıl ortaya çıktığı belli değil. Tüm vakaların yaklaşık yüzde 30'unda hastalık ailelerde görülür: etkilenenler mutasyonu bir veya iki ebeveynden miras almıştır.
Şimdiye kadar, Kallmann sendromuna neden olan birkaç mutasyon tanımlanmıştır. Bunlara KAL1, FGFR1, FGF8, CHD7, SOX10, PROKR2 ve PROK2 isimli mutasyonlar dahildir.
Araştırma ve teşhis
Semptomlar genellikle sadece ergenlik döneminde belirginleştiğinden, Kallmann sendromunun teşhisi birçok vakada geç konur. Bu, özellikle cinsel gelişim eksikliğinin genellikle daha açık bir şekilde görüldüğü erkek çocuklar için geçerlidir. Kızlarda semptomlar genellikle daha az belirgindir, bu nedenle sadece ilk adet döneminin olmaması bir doktor ziyaretine yol açar. Ek olarak, çoğu durumda, etkilenenler semptomların arkasında bir hastalıktan şüphelenmezler, bunun yerine "geç geliştiklerine" inanırlar.
Bazı durumlarda, hastalık bebeklerde zaten belirgindir: Etkilenen erkek çocuklarda inmemiş testisler (kriptorşidizm) ve/veya çok küçük bir penis (mikropenis) olabilir.
KS sendromunun herhangi bir belirtisi varsa ilk temas noktası çocuk doktoru, istemsiz çocuksuzluk durumunda jinekolog veya ürologdur.
Doktor aşağıdaki muayeneleri yapar:
Aile öyküsü: Kallmann sendromundan şüpheleniliyorsa, doktor ailede bilinen herhangi bir KS vakası olup olmadığını sorar.
Fizik muayene: Yaşına göre çok küçük bir penis veya ilk adet döneminin olmaması gibi sekonder cinsel özelliklerin gelişmemiş olması doktora KS sendromu hakkında ilk ipuçlarını verir. Ayrıca vücut büyüklüğüne, koltuk altı, göğüs ve kasık kıllarına da dikkat eder ve sakal büyümesini değerlendirir.
Koku testi: Bu test yaklaşık beş yaşından itibaren yapılabilir. Doktor, hastanın kokuyu algılayıp algılamadığını kontrol etmek için vanilin gibi saf kokular kullanır.
Kan testi: Doktor kan testi ile değişen hormon seviyesini belirleyecektir. Tipik olarak GnRH, LH ve FSH seviyeleri azalır veya düşük-normal bir seviyededir. Ergenlik çağındaki kız ve erkeklerde seks hormonlarının seviyeleri ergenlik öncesidir.
Testislerin ultrason muayenesi: Doktor, testisler gibi yumuşak dokuları incelemek için ultrason kullanır.
Bilgisayarlı tomografi (BT) veya manyetik rezonans tomografi (MRT): Bu muayene işlemleri ile doktor beyindeki koku alma merkezinin (olfactory bulb) gelişip gelişmediğini kontrol eder.
Elin röntgen muayenesi: Elin röntgen muayenesi yardımıyla doktor, büyüme plakalarının zaten kapalı olup olmadığını ve böylece vücudun büyümesinin bitip bitmediğini belirler.
Spermiyogram: Doktor menide sperm olup olmadığını inceler.
Genetik test: Genetik test, hastalığa neden olan kesin mutasyonu belirlemek için kullanılır.
Kallmann sendromu nasıl tedavi edilir?
Kallmann sendromu kolayca tedavi edilebilir. Etkilenenler seks hormonları ile replasman tedavisi alırlar. Hastalık ergenlikten önce tanınır ve tedavi edilirse, etkilenenler genellikle büyük ölçüde sınırsız bir yaşam sürerler. Buna düzenli ergenlik gelişimi ve normal bir cinsel yaşam dahildir.
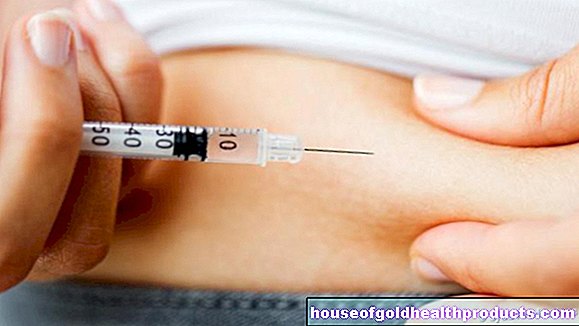
Cinsiyet hormonu uygulaması: Erkeklere testosteron, kadınlara östrojen ve progesteron verilir. Hormon preparatları enjeksiyonlar, jeller veya yamalar şeklinde mevcuttur. Hormon tedavisi genellikle erkeklerde yaşam boyu, kadınlarda ise menopoza kadar devam eder.
Vakaların yüzde 10 ila 20'sinde, hormon replasman tedavisi sona erdikten sonra doğuştan gelen bir GnRH eksikliği düzelir. Tedaviyi takiben hastalar normal hormon seviyelerine sahiptir ve normal cinsel olgunluk yaşarlar. Bu nedenle doktorlar, tedaviye devam etme ihtiyacını belirlemek için her bir ila iki yılda bir tedaviyi duraklatmanızı önerir.
Çocuk sahibi olmak istiyorsanız seks hormonu vermek: Spermin oluşması için vücudun GnRH'ye ihtiyacı vardır. Bu nedenle baba olmak isteyen erkeklere testosteron yerine GnRH hormonu verilir. Sperm üretiminin başlaması 18 ila 24 ay sürer. Vakaların yaklaşık yüzde 80'inde, erkekler daha sonra doğurgandır. İnmemiş testisli erkeklerin prognozu biraz daha düşüktür.
Osteoporoz tedavisi: Kemik yoğunluğu azalmış hastalar kalsiyum ve D vitamini alırlar. Ayrıca kemikleri güçlendirmek için spor ve egzersiz önerilir.
Anosmi: Şu anda koku duyusunu geri kazandıracak bir tedavi yoktur.
Psikoterapi: Bazı KS hastaları için hastalık ciddi bir psikolojik yüktür. Psikoterapist burada doğru kişidir.
Hastalığın seyri ve prognozu
Kallmann sendromunun nasıl ilerlediği kişiden kişiye değişir. Semptomlar hastadan hastaya değişir. Hastalığın bebeklik döneminde fark edilmesi, ergenlik döneminde teşhis edilmesi veya hormon eksikliğinin sadece yetişkinlikte ortaya çıkması olasıdır.
Hastalık zamanında teşhis edilirse prognozu çok iyidir. Uygun hormonal tedavi ile tüm hastalarda cinsel olgunlaşma sağlanır. Hastalar düzenli bir puberte gelişimi yaşarlar, doğurgandırlar ve normal bir yaşam beklentisine sahiptirler. Çocuk sahibi olmak isteyen hastaların hemen hepsi uygun tedavi ile fertil olurlar.
KS ancak 16 yaşından sonra tanınır ve tedavi edilirse, zaten uzamış olabilir. Bu değişiklik ilaçla bile geri döndürülemez.
Önlemek
Genetik bir hastalık olduğu için önlenmesi mümkün değildir.
Etiketler: parazitler gpp spor fitness

.jpg)