Omuriliğe bağlı kas atrofisi
ve Florian Tiefenböck, doktor Tarihinde güncellendiMaximilian Reindl, Münih'teki LMU'da kimya ve biyokimya okudu ve Aralık 2020'den beri editör ekibinin bir üyesi. Tıbbi, bilimsel ve sağlık politikası konularını sizin için anlaşılır ve anlaşılır kılmak için tanıyacaktır.
Maximilian Reindl'ın diğer gönderileriFlorian Tiefenböck, Münih LMU'da insan tıbbı okudu. Mart 2014'te'a öğrenci olarak katıldı ve o zamandan beri editör ekibine tıbbi makalelerle destek verdi. Augsburg Üniversite Hastanesi'nde tıp lisansını ve dahiliye alanında pratik çalışmasını aldıktan sonra, Aralık 2019'dan beri ekibinin daimi üyesidir ve diğer şeylerin yanı sıra araçlarının tıbbi kalitesini sağlar.
Florian Tiefenböck'ün diğer gönderileri Tüm içeriği tıp gazetecileri tarafından kontrol edilir.
Spinal müsküler atrofi veya kısaca SMA, omurilikteki belirli sinir hücrelerinin öldüğü nadir bir hastalıktır. Beyinden gelen uyaranlar ve dürtüler artık hedeflerine ulaşmaz: kaslar. Bu kas erimesine ve felce neden olur. SMA'nın farklı biçimleri vardır. En şiddetlisi bebeklik döneminde başlar. Yeni tedaviler, sağlıkta kalıcı bir iyileşme vaat ediyor. Spinal müsküler atrofiler hakkında daha fazla bilgiyi buradan okuyun.
Bu hastalık için ICD kodları: ICD kodları, tıbbi teşhisler için uluslararası kabul görmüş kodlardır. Örneğin, doktor mektuplarında veya iş göremezlik belgelerinde bulunabilirler. G12

Kısa bir bakış
- Spinal müsküler atrofi nedir? Bir grup kas zayıflığı hastalığı. Omurilikte kasları kontrol eden belirli sinir hücrelerinin (motor nöronlar) ölümüne dayanırlar. Bu nedenle SMA motor nöron hastalıkları arasındadır.
- Hangi formlar var? Kalıtsal spinal müsküler atrofiler çoğunlukla SMA'dır ve kromozom 5 (5q ile ilişkili SMA) üzerinde belirli bir genetik kusur vardır. Doktorlar dört farklı formu ayırt eder: SMA tip 1 - tip 4. Ayrıca, kalıtımı kesin olmayan sporadik formlar vardır.
- Sıklık: Nadir Hastalık; kalıtsal SMA, 7000'de yaklaşık bir yenidoğanı etkiler.
- Semptomlar: kas seğirmesi, ilerleyici kas zayıflığı, kas kaybı, felç. Gradyanlar, SMA şekline bağlı olarak farklılık gösterir.
- Nedenleri: Kalıtsal spinal müsküler atrofi tipleri 1-4, kromozom 5 üzerindeki, daha kesin olarak SMN1 genindeki genetik bir kusurun sonucudur. Sonuç olarak, vücut özel bir protein olan SMN proteininden yoksundur. Bu eksiklik omurilikteki motor nöronlara zarar verir.
- Teşhis: Değiştirilmiş SMN genetik yapısı için genetik muayene, fizik muayene, elektronörografi, elektromiyografi, kan testleri (örn. CK)
- Tedavi: Gen değiştirme tedavisi veya ekleme modülatörlerinin ilaç uygulaması mümkündür. Eşlik eden fizyoterapi, konuşma terapisi, ağrı tedavisi ve psikoterapi. Gerekirse, omurgada ameliyat. SMA tipine göre tedavi planı.
- Prognoz: Kalıtsal proksimal SMA durumunda, yeni tedavi seçenekleri nedensel bir etkiye sahiptir ve hastalığın seyri üzerinde olumlu bir etkisi olabilir. Tedaviye erken başlamak çok önemlidir. Tedaviler henüz her hasta için mevcut değildir. SMA tip 1'li çocuklar tedavi edilmezse genellikle ilk iki yıl içinde ölürler. Tip 3 ve tip 4'te yaşam beklentisi çok az veya hiç azalmadı.
Spinal müsküler atrofi nedir?
Spinal müsküler atrofide (SMA), omurilikteki belirli sinir hücreleri ölür. Genellikle kasları kontrol ederler, bu yüzden uzmanlar bu sinir hücrelerini motor nöronlar olarak adlandırır. Buna göre, SMA sözde motor nöron hastalıklarına aittir.
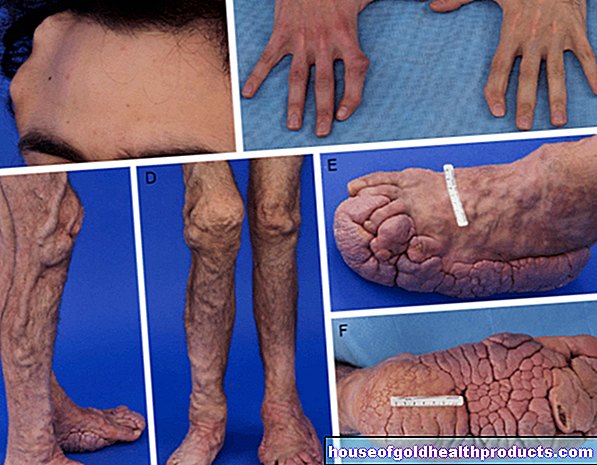
Spinal müsküler atrofilerde, uzantıları ile doğrudan kaslara bağlı olan alt (ikinci) motor nöronlar etkilenir. Hasar sonucunda kaslara daha az veya daha fazla sinir sinyali ulaşır. Kaslar giderek zayıflar ve küçülür (kas erimesi/kas atrofisi).
Doktorlar, farklı spinal müsküler atrofi formları arasında ayrım yapar. Açık farkla en büyük grup, gövdeye yakın (proksimal) kasların etkilendiği kalıtsal SMA'dır. Belirli bir genetik kusura dayanırlar. Yaklaşık 7.000 yenidoğandan biri onu geliştirecek.
Spinal müsküler atrofi genel olarak nadir görülen bir hastalıktır. Yine de en sık görülen ikinci otozomal resesif kalıtsal hastalıktır ve aynı zamanda genetik bir bozukluğa bağlı bebeklerde veya küçük çocuklarda en sık ölüm nedenidir.
Ne tür spinal müsküler atrofi vardır?
Doktorlar, kalıtsal (kalıtsal) SMA formlarını sporadik formlardan ayırır. Spinal müsküler atrofinin başka bir sınıflandırması, öncelikle ilk etkilenen kas gruplarıyla ilgilidir. Var
- Proksimal SMA: Yaklaşık yüzde 90 ile en büyük SMA grubunu oluştururlar. Semptomlar gövdeye yakın kaslarda yani proksimalde başlar.
- Proksimal olmayan SMA: Burada önce eller ve ayaklar gibi daha uzak kas grupları etkilenir (distal SMA). İlerleyen süreçte, bu SMA vücudun ortasına yakın kaslara da yayılabilir.
- Özel formlar (örn. Kennedy tipi spinobulbar müsküler atrofi)
Proksimal spinal müsküler atrofiler
Kalıtsal proksimal spinal müsküler atrofiler çoğunlukla spesifik bir genetik kusura dayanan hastalıklardır (5q ile ilişkili SMA, kromozom 5'te kusur). Bunlar sırayla dört farklı forma ayrılır. Sınıflandırma, ilk semptomların ortaya çıktığı zamana ve hastalığın seyrine dayanmaktadır.
Spinal Musküler Atrofi Tip 1: Bu, SMA'nın en yaygın ve en ciddi şeklidir. Aynı zamanda "Werdnig-Hoffmann hastalığı" veya "akut infantil SMA" olarak da adlandırılır. Hastalık genellikle erken bebeklik döneminde başlar. Kas zayıflığı tüm vücudu etkiler - doktorlar ayrıca bir "disket bebek sendromundan" bahseder (İngilizceden sarkık = sarkık, bebek = bebek, çocuk). SMA tip 1'li tedavi edilmeyen çocukların çoğu, iki yaşına gelmeden ölmektedir.
Spinal müsküler atrofi tip 2: Bu SMA formu, "orta spinal müsküler atrofi" veya "kronik infantil SMA" olarak da bilinir. İlk belirtiler tipik olarak 18 aylıktan önce ortaya çıkar. Etkilenenler bazen önemli ölçüde azaltılmış bir yaşam beklentisine sahiptir.
Spinal müsküler atrofi tip 3: "Juvenil spinal müsküler atrofi" veya "Kugelberg-Welander hastalığı" olarak da bilinir. Bu SMA genellikle 18 aylıktan sonra ve erken yetişkinlikten önce başlar. Kas zayıflığı tip 1 veya 2'den daha hafiftir. Etkilenenlerin yaşam beklentisi sadece biraz azalır.
Spinal müsküler atrofi tip 4: SMA tip 3'e benzer, ancak sadece yetişkinlikte (genellikle> 30 yaş) ortaya çıkar. Bununla birlikte, kas zayıflığı SMA tip 3'e göre daha az belirgindir ve daha yavaş ilerler.
Farklı versiyonlar arasındaki geçişler akıcıdır. Bazı durumlarda, bu net bir sınır çizmeyi zorlaştırır. Bazı genetik yatkınlıklar da söz konusu hastalığın şiddetinde önemli rol oynamaktadır.
Diğer spinal müsküler atrofiler
Bu proksimal formlara ek olarak, başka spinal müsküler atrofi formları da vardır. Bunlar, örneğin, daha nadir, ayrıca kalıtsal distal spinal müsküler atrofileri içerir. Onlarda semptomlar tipik olarak vücuttan daha uzaktaki kas gruplarında başlar.
Sporadik SMA durumunda kalıtım garanti edilmez. Ayrıca herhangi bir ailesel birikim tespit edilememektedir. Literatürde bunlar şunları içerir:
- Hirayama tipi (juvenil distal SMA, 15 yaş civarındaki hastalık, kol kaslarını etkiler, genellikle tedavi olmaksızın durur ve hatta iyileşebilir)
- Vulpian-Bernhard tipi (genellikle 40 yaşından itibaren omuz kuşağında başlayan "yelken kol" sendromu)
- Duchenne-Aran tipi (başlangıçta etkilenen el kasları, genellikle 30 yaşından sonra gövdeye doğru yayılır)
- Peroneal tip (önce alt bacak kaslarında görülen "yelken bacak" sendromu)
- Progresif bulbar palsi (konuşma ve yutma bozuklukları, amyotrofik lateral sklerozlu hastaların yaklaşık yüzde 20'sini etkiler)
Bazı sporadik SMA formları ("yelken kol - / - bacak" sendromu, progresif bulbar paralizi), uzman çevrelerde amyotrofik lateral sklerozun (ALS) varyantları arasında sayılır. Bu makale öncelikle kalıtsal proksimal spinal müsküler atrofilerle ilgilidir.
Spinobulbar müsküler atrofi
Spinobulbar veya bulbospinal müsküler atrofi (Kennedy tipi, Kennedy sendromu) kalıtsal bir hastalıktır. Genellikle genç ve orta yetişkinlik döneminde başlar. Bu özel SMA formu, X'e bağlı çekinik bir şekilde kalıtılır ve bu nedenle yalnızca erkekleri etkiler (erkeklerde yalnızca bir X kromozomu olduğundan, kadınlarda ikinci, sağlıklı X kromozomu baskındır ve kusuru telafi eder).
Bacaklarda ve kollarda veya omuzlarda vücuda yakın kaslarda, dil ve boğaz kaslarında kas güçsüzlüğü yaygındır. Sonuç olarak, etkilenenler örneğin konuşma ve yutma sorunları yaşarlar. Ayrıca titreme, kas krampları ve seğirmeden şikayet ederler. Etkilenen erkekler de genellikle bodur testislere sahiptir ve kısırdır. Ayrıca meme bezleri büyür (jinekomasti).
Spinobulbar müsküler atrofi genellikle yavaştır. Yaşam beklentisi pek kısıtlı değildir.
Spinal müsküler atrofiyi nasıl tanırsınız?
Tipik spinal müsküler atrofi, felce (parezi) kadar ilerleyici kas zayıflığı ve kas seğirmesidir. Sinir hasarının bir sonucu olarak, kaslar artık elektriksel uyarıları almaz, bu da zamanla küçülmelerine (kas atrofisi) neden olur. Kesin işaretler ve şikayetler ilgili forma bağlıdır. Aşağıdaki bölüm, kalıtsal proksimal SMA'nın semptomlarına bakar.
İnfantil Spinal Musküler Atrofi Tip 1 Belirtileri
SMA tip 1 ile semptomlar yaşamın ilk altı ayında ortaya çıkar. Genelleştirilmiş kas zayıflığı oluşur - yani tüm vücudu etkiler. Ayrıca kaslar arasındaki gerilim azalır. Doktorlar hipotoniden bahseder.
Yenidoğanlarda bu kas zayıflığı başlangıçta kendini yalancı bir kurbağayı (kurbağa bacağı duruşu) andıran tipik bir bacak duruşunda gösterir. Bacaklar bükülü, dizler dışa açılı ve ayaklar içe açılıdır. Başınızı kendi başınıza kaldırmak veya tutmak bile genellikle mümkün değildir.
İleri yaşta, SMA tip 1 olan çocuklar kendi başlarına oturamaz veya yürüyemezler. Dil kasları da etkilenebileceğinden birçok çocuk konuşamaz.
Tip 1 spinal müsküler atrofinin bir başka özelliği de üst vücudun şeklidir: göğüs ve sırt kasları düzgün gelişmez. Bu, üst gövdeye çan benzeri bir şekil verir (çan göğsü). Göğüs ve sırttaki kasların düşük gelişimi nedeniyle, etkilenenler kambur bir duruş benimser.
Genellikle omurganın artan bir eğriliği de vardır (skolyoz). Öne kambur ve kambur duruş, daha fazla solunum problemine neden olur. Çok hızlı ve sığ solunum (taşipne) karakteristiktir.
Orta spinal müsküler atrofi tip 2 belirtileri
Tip 2 spinal müsküler atrofi genellikle sadece yedi ila 18 aylık arasında semptomlara neden olur. Etkilenen çocuklar kendi başlarına oturabilirler, ancak genellikle ne ayakta durmayı ne de yürümeyi öğrenirler. Kas zayıflığı tip 1'den daha yavaş ilerler.
SMA tip 2 ile, omurganın deformasyonu gibi şiddetli infantil formunkine benzer semptomlar zamanla ortaya çıkar. Eklemler kısalan kaslar ve tendonlar (kontraktürler) nedeniyle sertleşir. Diğer belirtiler, ellerde titreme ve dildeki kasların seğirmesidir.
Juvenil Spinal Musküler Atrofi Tip 3 Belirtileri
Tip 3 spinal müsküler atrofi tipik olarak 18 aylıktan sonra ve 18 yaşından önce ortaya çıkar. Etkilenen çocuklar bağımsız olarak oturabilir, ayakta durabilir ve yürüyebilir. Ancak özellikle pelvik ve bacak kaslarındaki kas güçsüzlüğü, sallanarak yürüyüşe neden olur.
Birkaç yıl boyunca performans düşer: İlk başta, etkilenenler spor aktivitelerini veya merdiven çıkmayı zor bulurlar, ancak sonunda örneğin alışveriş çantalarını taşımak da zordur. Uzun yıllar sonra tip 3 spinal müsküler atrofi, yaşlı insanlarda bile koşmayı ve diğer herhangi bir eforu zorlaştırır, hatta imkansız hale getirir.
Bununla birlikte, genel olarak, semptomlar hastalığın diğer iki türünden, tip 1 ve tip 2'den daha az belirgindir. Etkilenenlerin çoğu için, yaşam kalitesi uzun bir süre boyunca neredeyse hiç bozulmaz.
Yetişkin spinal müsküler atrofi tip 4 belirtileri
Bu çok nadir görülen ilerleyici kas kaybı biçimi, yetişkinlikte, genellikle yaşamın üçüncü on yılında başlar. Bacak ve kalça kasları başlangıçta etkilenir. Hastalık ilerledikçe kas güçsüzlüğü omuzlara ve kollara da yayılır.
Klinik tablonun tezahürü jüvenil SMA tip 3'ünkine benzerdir. Bununla birlikte, progresif kas zayıflığı SMA tip 3'ten bile daha yavaştır.
Spinal müsküler atrofiye ne sebep olur?
Spinal müsküler atrofide, omurilikteki ikinci motor nöronlar yok olur. Bunlar, uzantıları ile kasları kontrol eden sinir hücreleridir. Bu son derece özelleşmiş motor nöronların hasarının bir sonucu olarak, sağlıklı insanlara göre daha az elektrik sinyali kaslara ulaşır. Kas hücreleri daha az kullanılırsa ve dolayısıyla daha az uyarılırsa, vücut onları zamanla parçalar.
genetik kusur
Çoğu durumda, spinal müsküler atrofi kalıtsal bir hastalıktır (kalıtsal SMA). Tipik proksimal SMA formlarının nedeni, hastanın genetik yapısındaki yanlış bilgidir. Kromozom 5 üzerindeki sözde SMN1 geni işlevsel değildir.
SMN1 geni, SMN adı verilen hayati protein molekülü için bilgiyi - yani planı - taşır. SMN, "Motor Nöronun Hayatta Kalması" anlamına gelir. SMN protein molekülü olmadan motor nöronlar zamanla yok olur.
Vücutta, prensipte işlevsel olmayan SMN1 genetik bilgisini “telafi edebilen” ilgili bir SMN2 geninin de olduğu doğrudur. Ancak bu genellikle sadece küçük bir ölçüde olur. Bu, SMN1 geninin bir işlev kaybının (tedavi edilmezse) genellikle bozulmamış bir SMN2 gen kopyası ile tamamen telafi edilemeyeceği anlamına gelir.
Otozomal çekinik ve otozomal dominant kalıtım
Bir kişinin genetik bilgisi iki kopya halinde mevcuttur. Sonuç olarak, herkes SMN1 geninin iki kopyasına sahiptir - biri babasından diğeri annesinden. Çocukluğun proksimal spinal müsküler atrofileri tipik olarak otozomal resesif bir özellik olarak kalıtılır.
Bu, spinal müsküler atrofinin yavrularda gelişmesi için ebeveynlerden gelen her iki gen varyantının (alel) kusurlu olması gerektiği anlamına gelir. Çekinik kalıtım durumunda, ebeveynler etkilenmez, çünkü işlevsiz olana ek olarak, kusuru telafi eden sağlıklı bir SMN1 genine de sahiptirler.
Yaklaşık her 45 kişiden biri SMA için bu sistemin sahibidir. Her iki partnerin de taşıyıcı olduğu bir çiftin bu hastalığa sahip bir çocuğa sahip olma riski %25'tir.
Ergenlikte birkaç vakada, özellikle erişkinlikteki spinal müsküler atrofiler de otozomal dominant geçişi takip eder. Baskın kalıtım durumunda, kusurlu bir gen zaten kendini gösterir - ve etkilenenler hastalanır. Ancak bu, kromozom 5 üzerinde yukarıda bahsedilen genetik kusur değildir. Bu 5q ile ilişkili SMA'lar her zaman otozomal çekinik bir şekilde kalıtılır.
Diğer SMA formlarıyla kalıtım
Proksimal olmayan spinal müsküler atrofi de kalıtsal olabilir. Özel spinobulbar formu (Kennedy tipi), bir cinsiyet kromozomu, X kromozomu yoluyla çekinik olarak kalıtılır (bu, erkek cinsiyet hormonları için yerleştirme bölgelerinin planını içeren gen varyantlarını etkiler). Sporadik formlarda ise kalıtım garanti edilmez. Burada tam olarak ikinci motor nöronların neden yok olduğu çok az biliniyor.
Muayeneler ve teşhis
Spinal müsküler atrofi teşhisi genellikle çocuk doktorları, sinir hastalıkları konusunda uzmanlaşmış çocuk doktorları (nöropdiatristler) ve sinir sistemi hastalıkları konusunda uzmanlar (nörologlar) tarafından yapılır. Daha kesin bir açıklama için çeşitli incelemeler gereklidir. Bir SMA durumunda, sinir ve kasların genetik testleri ve muayeneleri özellikle önemlidir.
Tıbbi geçmişin toplanması (anamnez)
Her hastalıkta doktor önce ortaya çıkan belirtileri ve şimdiye kadar nasıl ilerlediğini sorar. Bebeklerde ve küçük çocuklarda ebeveynler, çocuklarının davranışlarındaki değişiklikleri ve anormallikleri bildirir. Özellikle kalıtsal hastalıklar söz konusu olduğunda, doktorlar ayrıca ailenin tıbbi geçmişine de odaklanır.
Fizik muayene
Temel olarak, bir doktor çocuğu fiziksel olarak inceleyerek motor gelişimdeki anormallikleri belirler. Örneğin, çocukların bağımsız olarak başlarını dik tutup oturamadıklarını, kollarını veya bacaklarını bağımsız olarak hareket ettirip ettiremediklerini (yaşlarına bağlı olarak) test eder.
Daha büyük çocuklarda ve spinal müsküler atrofi şüphesi olan erişkinlerde tamamlayıcı egzersiz testleri yapılır. Doktor, ilgili kişinin ne kadar güç toplayabileceğini ve ne kadar süre tutabileceğini kontrol eder. Ayrıca dayanıklılığı da inceler.
Ek olarak, doktor, özellikle belirgin spinal müsküler atrofiler durumunda, tipik olarak zayıflayan veya sönen refleksleri test eder. Bunu yapmak için, örneğin topuk veya diz altı gibi çeşitli tendonlara çekiçle vurur ve reaksiyonu kontrol eder.
Genetik Çalışmalar
(Kalıtsal) spinal müsküler atrofi için en güvenilir tespit yöntemi genetik analizdir.Doktorlar, değiştirilmiş (mutasyona uğramış) bir SMN1 geninin kanıtını ve mevcut SMN2 kopyalarının sayısını ararlar.
Genel kural (kalıtsal) SMA'yı mümkün olduğunca erken teşhis etmek ve tedavi etmektir. Forma ve mevcut tedaviye bağlı olarak, omuriliğin motor nöronları geri döndürülemez şekilde hasar görmeden önce motor gelişim olumlu etkilenebilir.
SMA'da daha fazla araştırma
Bir SMA'dan şüpheleniliyorsa, doktorlar genellikle sinirlerin iletim hızını (elektronörografi) ve kas aktivitesini (elektromiyografi) ölçer. Gerekirse ultrason (miyosonografi) veya manyetik rezonans görüntüleme (MRI) kullanarak kasları da incelerler.
Ek olarak, doktorlar kan testleri düzenler. Spinal müsküler atrofi varsa, belirli parametreler değiştirilebilir: örneğin, kreatin kinaz (CK, tipik bir kas enzimi) düzeyi yükselir.
Spinal müsküler atrofilerin tedavisi
Spinal müsküler atrofinin tedavisi karmaşıktır. Uzun bir süre boyunca, herhangi bir SMA formu için nedensel tedavi mümkün değildi. Bununla birlikte, tıbbi araştırmalardaki ilerlemeler, doktorlara, proksimal SMA'dan (kromozom 5'te SMN gen defekti) etkilenenlere temel olarak yardımcı olmak için yeni tedavi seçenekleri sunmaktadır.
Ek olarak, doktorlar semptomları hafifletmeye ve etkilenenlere mümkün olan en iyi desteği sağlamaya odaklanır (örneğin, fizyoterapi, solunum tedavisi, psikoterapi, muhtemelen cerrahi).
Tıbbi terapi
SMA'nın bilinen bir SMN gen kusuruna dayandığı hastalar için yeni tedavi yaklaşımları, doğrudan genetik materyalin kendisine veya genetik bilginin aşağı yönde işlenmesine müdahale eder.
Amaç, hastanın vücudunun motor nöronlar için çok önemli olan yeterli miktarda SMN proteinini bağımsız olarak üretmesini sağlamaktır.
Spinal müsküler atrofi için aşağıdaki tedavi seçenekleri mevcuttur:
- Ekleme modülatörleri (Nusinersen, Risdiplam): Bu ilaçlar, haberci RNA moleküllerinin daha sonraki işlenmesine doğrudan müdahale eder. Sağlam SMN2 geninden daha yüksek miktarda SMN proteini sağlayan süreçleri güçlendirirler.
- Gen replasman tedavisi (Onasemnogene Abeparvovec): Bu terapi doğrudan insan genomuna müdahale eder. SMN1 geninin hatalı kopyası, etkilenen hücrelerde dışarıdan sağlanan, fonksiyonel bir gen yapısı ile değiştirilir.
Ekleme modülatörleri
Bir SMN1 gen kusuru durumunda, vücut alternatif olarak ilgili SMN2 geninden SMN proteini üretebilir. Yedek gen SMN2 "atlar", ancak bu yeterli değil. Nedeni: SMN2'deki proteinler genellikle çok kısadır ve hızla parçalanır.
Bu, karşılık gelen SMN2 haberci RNA'nın (SMN2 mRNA) işlenmesinden kaynaklanmaktadır. Yapı bilgisini genomdan (DNA) protein üretim bölgelerine (ribozomlar) iletir.
Bunu yapmak için önce genomdaki SMN2 geni okunur. Bir ön SMN2 haberci RNA üretilir. Diğer şeylerin yanı sıra, sözde ekleme yoluyla daha fazla işlenmelidir. Ancak o zaman olgun haberci RNA ortaya çıkar. Özel hücre kompleksleri, ribozomlar, daha sonra olgun haberci RNA'yı okur ve böylece SMN2 proteini üretir. Ve tam olarak kısaltılmış ve kararsız olan, hızla sökülen ve SMN1'in işlevini üstlenemeyen budur.
Bunu değiştirmek için aktif bileşenler Nusinersen ve Risdiplam, ön haberci RNA'nın daha sonraki işlenmesini etkiler. Sonuç olarak, bu sözde ekleme modülatörleri, sonuçta kullanılabilir SMN proteinlerinin miktarını arttırır ve böylece yeterli bir tedarik sağlayabilir.
Nusinersen
Nusinersen ilacı, "antisens oligonükleotid" (ASO) olarak adlandırılan bir ilaçtır. 2017 yılında Avrupa İlaç Ajansı tarafından onaylanmıştır. ASO, yapay olarak üretilmiş ve özel olarak uyarlanmış RNA molekülleridir. SMN2 haberci RNA'ya spesifik ve kesin olarak bağlanırlar. Bu, insan hücresinde daha fazla yanlış işlenmelerini önler.
Spesifik olarak: Nusinersen, önemli bilgilerin (ekson 7) SMN2 haberci RNA'sından yanlışlıkla kesilmesini önler. Ekson 7'nin nerede olduğu, vücudun daha sonra daha işlevsel SMN proteini yapmasına neden olur.
Nusinersen, lomber ponksiyon olarak bilinen yolla verilir. Bu, ilacın bir şırınga ile spinal kanala enjekte edilmesi anlamına gelir. Bu terapi birkaç aylık düzenli aralıklarla tekrarlanır. Tedavinin ilk yılında, etkilenenler yılda altı, ardından üç doz alırlar.
Hastalar genellikle ilacı iyi tolere eder. Nusinersen daha uygun hastalık seyrine yol açar. Çalışmalar birçok hastada hareketliliğin arttığını göstermiştir: birçok durumda serbestçe oturmak ve vücudu bağımsız olarak döndürmek mümkün olmuştur. Yan etkiler ve komplikasyonlar, diğer şeylerin yanı sıra, lomber ponksiyona dayanır (örn. baş ağrısı, meninks enfeksiyonları).
risdiplam
Avrupa Komisyonu, Mart 2021'de Risdiplam'ı 5q ile ilişkili SMA'ya (tip 1-3 veya bir ila dört SMN2 gen kopyası) karşı üçüncü ilaç olarak onayladı. Risdiplam günlük olarak çözünmüş bir toz olarak alınır. Kesin doz, yaşa ve vücut ağırlığına göre hesaplanır.
Nusinersen'den farklı olarak, Risdiplam bir "antisens oligonükleotid" değil, küçük bir moleküldür. Bu molekül, SMN2 proteinleri için haberci RNA'ya bağlanır ve onları bu şekilde stabilize eder. Sonuç olarak, daha işlevsel SMN proteinleri oluşturulur.
Risdiplam'ın yaygın yan etkileri arasında gastrointestinal rahatsızlık, kızarıklık, ateş ve idrar yolu enfeksiyonları bulunur.
Gen değiştirme tedavisi
Proksimal spinal müsküler atrofinin tedavisine yönelik başka bir yaklaşım, gen replasman tedavisi olarak bilinen şeye dayanır. Arızalı SMN1 geni - ilerleyici SMA'nın başlangıç noktası - genin yeni bir işlevsel kopyası ile "yerini alır".
Bu prensipte çalışan etken madde Onasemnogene Abeparvovec (AVXS-101), Mayıs 2020'de Avrupa İlaç Ajansı'ndan (EMA) yeni yürümeye başlayan çocukların ve çocukların tedavisi için şartlı pazarlama izni aldı.
İlaç, EMA bilgisine göre SMA tip 1 için kullanılabilir. SMA hastalığının diğer tüm formlarında, genetik özellikler (SMN2 kopyalarının sayısı) gen replasman tedavisinin bir seçenek olup olmadığına karar verir.
Onasemnogene Abeparvovec ile insan SMN1 geninin işlevsel bir kopyası, omuriliğin ve beyin sapının etkilenen hücrelerine verilir. Bu, adeno-ilişkili viral vektörler (AAV vektörleri) olarak adlandırılan yeni genetik materyal için “feribot” görevi gören belirli virüsler tarafından yapılır.
Vektör gen yapıları bir kez damar yoluyla kan dolaşımına bir infüzyon olarak verilir ve oradan tüm vücuda dağıtılır. Küçük çocuklarda henüz tam olarak gelişmemiş kan-beyin bariyeri nedeniyle bu vektörler omurilik dokusuna da girebilir.
Tercihen bu vektörleri motor nöronların özel yüzey yapılarına bağlayarak, daha sonra SMN proteinini bağımsız olarak üretmek için tercihen genetik materyali alırlar.
Tedavi motor fonksiyonları iyileştirebilir ve kalıcı gelişimsel başarıya yol açabilir (örneğin oturma, emekleme ve desteksiz yürüme).Tedavi sırasında karaciğer değerleri bazen önemli ölçüde yükselebilir ancak kandaki trombosit sayısı düşebilir. Ateş ve kusma da yaygındır. Yan etkileri azaltmak için hastalara birkaç hafta kortikosteroidler ("kortizon") verilir.
Yaşa uygun motor gelişim genellikle ancak gen tedavisinin semptomatik olarak başlamasıyla mümkündür. Tedavi uzmanlaşmış nöromüsküler tedavi merkezlerinde gerçekleştirilir.
fizik Tedavi
Fizyoterapi, SMA tedavisinin önemli bir ayağı olmaya devam ediyor.Yeni tedavi yaklaşımları ile her SMA formu tedavi edilemez. Düzenli egzersiz tedavisi, fiziksel yetenekleri korumak ve kas yıkımını yavaşlatmak için tasarlanmıştır.
Fizyoterapist, vücudun zaten felçli olan kısımlarını pasif olarak hareket ettirir. Aktif hareket dizileri ise kasların hareketliliğini ve gücünü desteklemek için eğitilir. Masaj veya sıcak ve soğuk tedaviler de yardımcı olabilir. Bunlar ayrıca gevşemeye ve belirli koşullar altında daha fazla dejenerasyonu yavaşlatmaya hizmet eder.
Konuşma terapisi
Bazı durumlarda, SMA konuşma ve yutma kaslarını etkiler. Sonra bir konuşma terapisi egzersizi yardımcı olur. Çocukları konuşmayı öğrenmeye teşvik eder. Daha yaşlı hastalarda bile, bu genellikle konuşmadaki bozulmayı yavaşlatabilir. Konuşma terapistleri ayrıca doğru yutmayı da eğitir.
Hem fizyoterapistler hem de konuşma terapistleri, hedefe yönelik solunum terapisinden etkilenenleri destekler.
Ağrı kesici tedavi
Ağrı tedavisi, özellikle hastalığın daha ileri evrelerinde önemli bir rol oynar. Doktorlar, etkilenenlerin acısını azaltmak için ağrı kesici ilaçlar kullanır.
ameliyat
Spinal müsküler atrofi, omurganın şiddetli eğriliğine (skolyoz) yol açabileceğinden, doktorlar bazen ameliyatı düşünür. Bunu yaparken, özellikle omurgayı sertleştirirler.
Bu, etkilenenlere (belirli) ek bir gövde stabilitesi sağlar, bu da sadece daha dik bir duruş sağlamakla kalmaz, aynı zamanda kemikleri ve eklemleri de korur. Bir spinal cerrahi ayrıca ilerleyici solunum problemlerine karşı da yardımcı olabilir.
psikoterapötik bakım
Spinal müsküler atrofi gibi nöromüsküler hastalıklar büyük psikolojik stresi temsil eder.Hastalar ve yakınları, psikoterapi liderliğindeki bireysel ve grup seanslarında tanıyı işler ve hastalıkla daha iyi başa çıkmak için stratejiler geliştirir.
Kendi kendine yardım grupları ve hasta temsilcileri de önemli destekler sunmaktadır. Etkilenenleri ve yakınlarını bir SMA hastalığının zorluklarıyla başa çıkmaları için bilgilendirir, tavsiyelerde bulunur ve destekler.
Spinal müsküler atrofilerden iyileşme şansı
Spinal müsküler atrofi varsa, prognoz öncelikle ilgili şekle bağlıdır. Semptomlar ne kadar geç ortaya çıkarsa, kurs o kadar iyi olur. Ek olarak, doktorlar spinal müsküler atrofiyi ne kadar erken teşhis ederse, motor nöronlar geri dönüşü olmayan bir şekilde hasar görmeden önce bile uygun tedavi önlemlerini o kadar erken başlatabilirler.
Ekleme modülatörleri ve gen replasman tedavisi yoluyla yeni tedavi seçenekleri, özellikle tedaviye (çok) erken başlandığında, proksimal SMA tedavisinde büyük potansiyele sahiptir. Bununla birlikte, güvenilir bir uzun vadeli prognoz için veriler hala beklemededir. Yalnızca daha ileri çalışmalar ve sıkı sıkıya bağlı ilaç güvenliği gözlemleri, önümüzdeki (aylar ve) yıllar için daha fazla kesinlik sağlayabilir. Daha yeni ilaçlarla, hastalığın uzun vadeli kontrolü ve hatta bir tedavi en azından düşünülebilir.
SMA tip 1 genellikle ciddi bir hastalıktır.SMA tip 1 gelişen çocukların (tedavi edilmeyen) yaşam beklentisi çok sınırlıdır. Tüm vücutta hızla artan kas güçsüzlüğü nefes almayı da etkiler. Sonuçlar akut pnömoni ve hatta solunum yetmezliğidir. Etkilenen çocuklar yaşamın ilk birkaç yılında ölürler.
SMA tip 2 için prognoz biraz daha iyidir. Yaşam beklentisi, hastalığın tam şiddetine bağlı olarak değişir: bazıları çocuklukta ölür, ancak çoğu genç erişkinliğe ulaşır. Er ya da geç - istenirse - daha şiddetli formlarda solunum desteklenmelidir. Etkilenen insanlar tekerlekli sandalye yardımıyla hareket halinde kalırlar.
SMA tip 3 ile prognoz önemli ölçüde daha iyidir - özellikle ilk belirtiler geç ortaya çıkarsa. Performans, birkaç yıl boyunca kademeli olarak bozulur. Yaşlılıkta tekerlekli sandalye veya hatta kalıcı bakım gerekebilir. Yaşam beklentisi, tip 3 spinal müsküler atrofi ile pek kısıtlanmaz.
Yetişkin spinal müsküler atrofi (tip 4) tip 3'ten bile daha yavaştır. İnsanlar genellikle normal bir yaşam beklentisine sahiptir.