Kore avcılığı
Ingrid Müller bir kimyager ve tıp gazetecisidir. On iki yıl boyunca 'nin genel yayın yönetmenliğini yaptı. Mart 2014'ten bu yana Focus Gesundheit, sağlık portalı ellviva.de, yayınevi live crossmedia ve rtv.de sağlık kanalı için serbest gazeteci ve yazar olarak çalışıyor.
houseofgoldhealthproducts uzmanları hakkında daha fazla bilgi Tüm içeriği tıp gazetecileri tarafından kontrol edilir.Huntington hastalığı (Huntington hastalığı, modası geçmiş: St. Vitus's Dance) kalıtsal bir beyin hastalığıdır. Huntington hastalığı olan kişilerde, beynin kas kontrolü ve psikolojik işlev için önemli olan alanları yavaş yavaş yok edilir. Sinir hücreleri yavaş yavaş yok olur. Nedeni hatalı bir gendir Huntington hastalığı hakkında daha fazla bilgi edinin.
Bu hastalık için ICD kodları: ICD kodları, tıbbi teşhisler için uluslararası kabul görmüş kodlardır. Örneğin, doktor mektuplarında veya iş göremezlik belgelerinde bulunabilirler. G10

Huntington hastalığı: açıklama
Huntington hastalığı, Batı Avrupa ve Kuzey Amerika'da 100.000 kişiden yaklaşık yedi ila on kişiyi etkileyen, beynin çok nadir görülen kalıtsal bir hastalığıdır. Almanya'da yaklaşık 8.000 kişi Huntington hastalığından muzdarip.
Huntington hastalığı her yaşta ortaya çıkabilir. Huntington hastalığının ilk belirtileri genellikle 35 ila 45 yaşları arasında ortaya çıkar, ancak erken çocukluk veya yaşlılıkta da ortaya çıkabilir. Bu aynı zamanda, hastalığa neden olan değişikliğin (mutasyon) genetik yapıdaki tam olarak nerede olduğuna da bağlıdır. Tipik semptomlar, demans dahil olmak üzere hareket bozuklukları ve karakterdeki değişikliklerdir.
Huntington koresi seyrini etkileme olanakları sınırlıdır. Sinir hücrelerini koruyan ve ilerleyici nöronal yıkımı durduran çok sayıda madde test edildi. Ancak şimdiye kadar, bu maddelerin hiçbiri hastalığın seyri üzerinde önemli bir etkiye sahip olmamıştır. Bununla birlikte, Huntington hastalığının semptomlarını azaltmaya yardımcı olabilecek bazı ilaçlar mevcuttur.
Huntington hastalığı olan kişilere ve sevdiklerine yardım eden destek grupları vardır.
"Huntington Hastalığı" adının nereden geldiği
Huntington'un koresi, hastalığı ilk kez 1872'de tanımlayan ABD'li doktor George Huntington'a kadar uzanıyor. Ayrıca Huntington hastalığının kalıtsal olduğunu fark etti. Adı Yunancadan geliyor - "choreia" = "dans". Huntington hastalığının birkaç adı vardır: Huntington hastalığı, Huntington hastalığı veya geçmişte St. Bunun nedeni, hastaların yavaş yavaş hareketlerinin kontrolünü kaybetmeleridir - bu da hafifçe bir dansı anımsatabilir.
Huntington hastalığı: belirtiler
Huntington hastalığı: erken evreler
Huntington hastalığı genellikle zihinsel sağlık sorunları gibi ilerleyen ve spesifik olmayan semptomlarla başlar. Birçok hasta daha sinirli, agresif, depresif veya çekingendir; diğerleri kendiliğindenlik kaybı veya artan bir endişe hissederler.
Huntington hastalığındaki hareket bozuklukları genellikle başın, ellerin, kolların, bacakların veya gövdenin ani, istemsiz hareketlerinden oluşur. Aşırı durumlarda, bu, hastalığa özgü bir sıçrama yürüyüşüne yol açabilir. Bu nedenle Huntington hastalığına geçmişte "Aziz Vitus'un Dansı" da deniyordu.
Huntington hastalığı olan kişiler genellikle bu abartılı ve istenmeyen hareketleri başlangıçta hareketlerine dahil edebilirler. Gözlemci için bu, abartılı jestler izlenimi yaratır. Çoğu durumda, etkilenenler başlangıçta hareket bozukluklarını bu şekilde fark etmezler.
Huntington Hastalığı: Sonraki Aşamalar
Huntington hastalığı ilerledikçe, dil ve boğaz kasları da giderek daha fazla etkilenir. Dil dalgalı görünüyor, sesler patlayarak söyleniyor. Yutma bozuklukları da mümkündür - o zaman etkilenenlerin gıda bileşenlerini yutma ve pnömoni geliştirme riski vardır.
Huntington hastalığı ilerledikçe, hastalar genellikle amansız bir şekilde zihinsel yetilerini kaybederler. Yaklaşık 15 yıl sonra, etkilenenlerin hemen hepsinde demans tespit edilebilir.
Huntington hastalığının son evrelerinde hastalar çoğunlukla yatalaktır ve tamamen başkalarının yardımına bağımlıdır.
Huntington hastalığı benzeri hastalıklar
Huntington hastalığına benzer semptomlar, kalıtsal olmayan başka nedenlerle de tetiklenebilir. Örnekler, hamilelik sırasında bulaşıcı hastalıkların veya hormonal değişikliklerin sonuçlarıdır (kore gravidarum). İlaçlar ve hormonal kontraseptifler (hap gibi) daha az yaygın tetikleyicilerdir. Bu aynı zamanda beynin belirli bölgelerini etkileyen felçler için de geçerlidir. Beyindeki dolaşım bozuklukları nedeniyle yaşlılıkta bile bir tür kore ortaya çıkabilir. Hipertiroidizm gibi hastalıkların da koreye yol açabileceğine inanılmaktadır. Ancak burada seyir ilerleyici değildir ve hareket bozuklukları genellikle tekrar geriler. Bu gibi durumlarda şiddetli psikolojik belirtiler atipiktir.
Huntington hastalığı: nedenleri ve risk faktörleri
Huntington hastalığına, beynin belirli bölgelerindeki sinir hücrelerinin genetik ölümü neden olur. Hastalık hem erkekleri hem de kadınları etkileyebilir, çünkü sorumlu gen cinsiyete bağlı bir şekilde kalıtsal değildir - yani X ve Y cinsiyet kromozomlarında değil, otozomlarda. Bunlar cinsiyeti belirlemeyen ve iki kopya halinde (biri anneden, biri babadan) bulunan kromozomlardır.
Bir ebeveynin kusurlu geni taşıyan herhangi bir çocuğunun kusurlu geni alma ve Huntington hastalığına yakalanma riski yüzde 50'dir. Uzmanlar otozomal dominant kalıtımdan bahsediyor. Dominant, iki çift kromozomdan sadece birinde karşılık gelen gen değiştirilse bile hastalığın patlak vermesi anlamına gelir.
Kalıtım - kromozom dörtte hata
Genetik materyalin moleküler alfabesi (deoksiribonükleik asit, kısa DNA veya DNA) dört nükleik asitten oluşur: adenin, timin, guanin ve sitozin. Bu dört harfin yeni kombinasyonları, kromozom adı verilen iplik benzeri yapılar şeklinde depolanan tüm genetik bilgiyi oluşturur.
Huntington hastalığında, dördüncü kromozom üzerindeki bir gen değiştirilir (Huntington geni olarak da adlandırılır). 1993 yılında tanımlanmıştır. Bu gen tarafından kodlanan protein düzgün çalışmıyor ve bu da sonunda Huntington hastalığının semptomlarına yol açıyor.
Sağlıklı insanlarda, üç nükleik asit sitozin-adenin-guanin, bu alanda dördüncü kromozom üzerinde 10 ila 30 kez tekrarlanır. 36'dan fazla sitozin-adenin-guanin tekrarı (CAG tekrarları veya CAG üçlüleri) ile Huntington hastalığı patlak verir. 30 ila 35 CAG tekrarı gri alan olarak kabul edilir.
Huntington hastalığı olanların yaklaşık yüzde bir ila üçünde Huntington hastalığı ile kan bağı bulunamaz. Ya tamamen yeni bir gen değişikliği, sözde yeni mutasyon. Veya HD hastasının bir ebeveyni 30 ila 35 tekrara sahipti ve hastalığa yakalanmadılar. Bir sonraki nesilde, tekrar sayısı genellikle artar. 36'dan fazla çocuk Huntington hastalığına yakalanacak.
Dördüncü kromozomda ne kadar çok CAG tekrarı sayılırsa, Huntington hastalığı o kadar erken patlak verir ve hastalık o kadar hızlı ilerler.
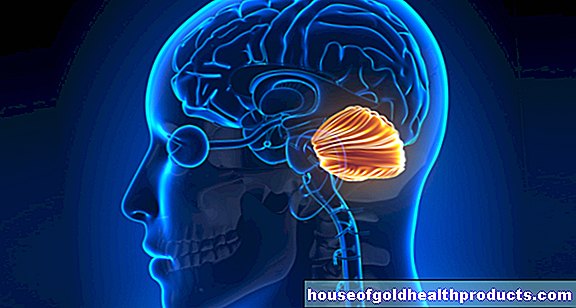
Huntington hastalığında sinir hücrelerinin ölümü serebral korteksi ve özellikle bazal gangliyonları etkiler. Bunlar, beynin iki yarısında yer alan ve diğer şeylerin yanı sıra koordineli hareket dizisinde rol oynayan büyük sinir hücresi gruplarıdır.
Huntington hastalığı: muayeneler ve teşhis
Huntington hastalığından şüpheleniliyorsa, uygun deneyimli nörologların bulunduğu uzmanlaşmış bir Huntington hastalığı merkezini görmek en iyisidir. Çünkü aile hekimleri ve bazen “normal” nörologlar bu hastalığa sahip bir hasta görmemiştir.
Huntington hastalığı tanısının başlangıcında aile öyküsü (anamnez) dahil olmak üzere ayrıntılı bir anket vardır. Yakın akrabaların (ebeveynler, büyükanne ve büyükbabalar) Huntington hastalığı olup olmadığı doktor için ilginçtir. Huntington hastalığının belirtileri (erken evrelerde başka hastalıkları da gösterebilir), hastalığın seyri ve nörolojik muayenedeki anormallikler de tanı için önemlidir.
Tanı kan testi ile doğrulanır. Dördüncü kromozom üzerindeki belirli bir gende aynı baz çiftlerinin (CAG) aşırı sayıda tekrarı moleküler genetik testlerle tespit edilebilir.
Genetik bir değişiklik bulunamazsa, başlangıçta Huntington hastalığına benzer semptomlar gösterebilecek hastalıkları dışlamak için tekrar kan alınır. Diğer şeylerin yanı sıra, kandaki tiroid değerleri ve belirli otoantikorların (vücudun kendi maddelerine yönelik antikorlar) konsantrasyonu belirlenir.
Sinir hasarını belirleyin
Sinir hasarının boyutunu belirlemek için Huntington hastalığı olan kişiler nörolojik, nöropsikolojik ve psikiyatrik olarak da incelenir. Bu muayeneler ya deneyimli doktorlar ya da özel eğitim almış nöropsikologlar tarafından yapılır.
Beynin bilgisayarlı tomografisi (BT) veya manyetik rezonans görüntülemesi (MRI) gibi görüntüleme incelemeleri, beynin tek tek alanlarının (örneğin kuyruk çekirdeği ve serebral korteks) dökümünü gösterebilir. Elektrofizyolojik teşhis, bireysel vakalarda Huntington hastalığı hakkında da önemli bilgiler sağlayabilir.
Huntington hastalığı için genetik testler
Huntington hastalığı riski yüksek olan sağlıklı insanlar bile genetik test yaptırabilir. Doktorlar tahmine dayalı teşhis veya tahmine dayalı teşhisten bahseder. Bu test, birisinin Huntington hastalığına yakalanıp yakalanmayacağını açıkça belirlemek için kullanılabilir.
Bununla birlikte, bu genetik değişimin taşıyıcısı olma bilgisi, etkilenenlerin ruhu üzerinde büyük bir etkiye sahip olabilir. Sonuç olarak, Huntington hastalığı için genetik test yaptırmak için yürürlükte olan yönergeler vardır. İlgili kişiler, diğer şeylerin yanı sıra, riskler hakkında önceden bilgilendirilmelidir. Küçükler üzerinde genetik test yapılamaz. Örneğin doktorlar, sigorta şirketleri, evlat edinme kurumları veya işverenler gibi üçüncü şahısların talebi üzerine kişiler söz konusu olduğunda bile.
Huntington hastalığı: tedavi
Huntington Hastalığı İlaçları
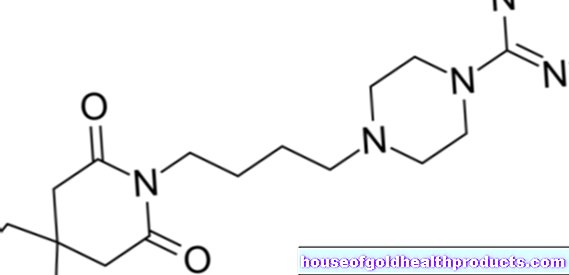
Huntington hastalığı nedensel olarak tedavi edilemez ve tedavi edilemez. Bununla birlikte, bazı ilaçlar semptomları hafifletir. Aktif maddeler tiapride ve tetrabenazin, vücudun kendi haberci maddesi dopamine karşı koyarak aşırı ve kontrolsüz hareketi kısıtlayabilir. Depresif ruh hallerinde, seçici serotonin geri alım inhibitörleri (SSRI) veya sülpirid grubundan antidepresanlar semptomları azaltabilir.
Huntington hastalığında demans gelişimine karşı resmi bir tedavi önerisi bulunmamaktadır. Bazı çalışmalarda, aktif bileşen memantin, zihinsel gerilemeyi yavaşlatmayı başardı.
İlaç bazlı Huntington hastalığı tedavisine ek olarak, fizyoterapi, uğraşı terapisi ve konuşma terapisi gibi destekleyici önlemler de semptomların hafifletilmesine yardımcı olabilir. Örneğin, konuşma veya yutma bozuklukları özel egzersizlerle iyileştirilebilir. Ayrıca, günlük yaşam aktivitelerini eğitmek için ergoterapi kullanılabilir. Bu, Huntington hastalığı olan kişilerin daha uzun süre bağımsız kalmalarını sağlar. Psikoterapi, birçok hastanın hastalıkla daha iyi başa çıkmasına yardımcı olur. Kendi kendine yardım grupları da birçokları için harika bir destek.
Huntington hastalığı olan birçok kişi hastalıkları boyunca önemli ölçüde kilo verdiğinden, yüksek kalorili bir diyet mantıklıdır. Bazı durumlarda, biraz fazla kilolu olmak Huntington hastalığının semptomlarını iyileştirir.
Huntington Hastalığı Araştırması
Huntington hastalığında altta yatan bozulma sürecini durdurabilecek tedaviler henüz bilinmemektedir. İlaç temelli Huntington hastalığı tedavisinde çeşitli yeni yaklaşımlar sürdürülmektedir, ancak bunlar henüz deneysel aşamadadır. Çalışmalar, yüksek dozlarda CoEnzyme Q10'un biraz olumlu etkileri olduğunu, ancak genel olarak hastalığın seyrini etkilemediğini göstermiştir. CoEnzyme Q10, vücutta doğal olarak oluşan ve hücreleri hasardan koruyan bir proteindir.
Huntington hastalığını tedavi etmek için kullanılan bir başka aktif bileşen, balık yağı bileşeni eikosapentaenoik asitten yapılan Huntington hastalığı için özel olarak geliştirilmiş bir ilaç olan etil ikosapent'tir. Ethyl-Icosapent, hücre ölümünde rol oynayan belirli hücre bileşenlerinin zarar görmesini önlemek ve hastalıklı sinir hücrelerinin etkinliğini artırmak için tasarlanmıştır. Bu ilaç da hareket bozukluklarında herhangi bir iyileşme sağlayamadı.
Huntington hastalığı: hastalık seyri ve prognoz
Huntington hastalığının tedavisi yoktur. Etkilenenler, Huntington hastalığının başlangıcından ortalama 19 yıl sonra ölmektedir. Bu yalnızca ortalama bir değerdir, bu da bireylerin Huntington hastalığının sonuçlarından çok daha sonra veya daha erken ölebileceği anlamına gelir.
Etkilenenlerin geliştirdiği semptomlar ve çeşitli ilaçlara ne kadar iyi yanıt verdikleri büyük ölçüde değişir. Yalnızca Huntington hastalığıyla günlük olarak ilgilenen uzmanlar, hastalığın birçok özelliğine yeterince aşinadır. Bu nedenle, bireysel tedaviyi hastalığın mevcut seyrine uyarlamak için HD hastalarının Almanya'daki Huntington hastalık merkezlerinden birinde kendilerini düzenli olarak kontrol ettirmeleri tavsiye edilir.
Huntington hastalığı olan kişilerin akrabaları ve yaşam partnerleri için hastalık duygusal, bazen de finansal olarak çok streslidir. Deutsche Huntington Hilfe e.V., gerekirse değerli tavsiyeler ve önemli adresler sağlayarak etkilenenlerin ailelerine açıktır.
Etiketler: stres önleme genç